Xiang-Ping Hu and Zhuo Zheng of the Dalian Institute of Chemical Physics
developed (Org. Lett. 2009, 11, 3226,
DOI: 10.1021/ol9012469;
J. Org. Chem. 2009, 74, 9191,
DOI: 10.1021/jo901619c)
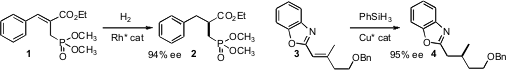
a family of Rh catalysts for the enantioselective hydrogenation of allylic
phosphonates such as 1. Hon Wai Lam of the University of Edinburgh established
(J. 3-Bromoquinolin-5-ol custom synthesis Am. PMID:25023702 Chem. Histamine Chemscene Soc. 2009, 131, 10386.
DOI: 10.1021/ja904365h)
that alkenyl heterocycles such as 3 could be reduced with high ee. The
product 4 could be hydrolyzed to the carboxylic acid, although that
transformation was not reported by the authors.
Ken Tanaka of the Tokyo University of Agriculture and Technology showed
(J. Am. Chem. Soc. 2009, 131, 12552.
DOI: 10.1021/ja905908z)
that an isopropenyl amide 6 could be hydroacylated with high ee. Gregory C. Fu of MIT observed
(J. Am. Chem. Soc. 2009, 131, 14231.
DOI: 10.1021/ja9061823)
that nitromethane 9 could be added to the allenyl amide 8 to give 10, the product of
γ-bond formation. Robert K. Boeckman, Jr. of the University of Rochester devised
(Org. Lett. 2009, 11, 4544.
DOI: 10.1021/ol9017479)
what appears to be a general protocol for the
construction of alkylated ternary and quaternary centers, enantioselective
hydroxymethylation of an aldehyde 11.
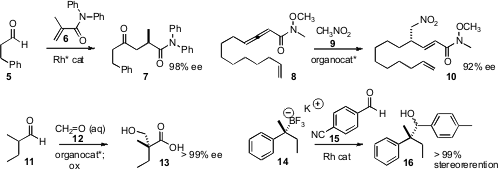
In another approach to the construction of alkylated quaternary centers,
Varinder K. Aggarwal of the University of Bristol demonstrated
(Angew. Chem. Int. Ed. 2009, 48, 6289.
DOI: 10.1002/anie.200901900)
that an enantiomerically-enriched trifluoroborate salt
14 could be added to an aromatic aldehyde 15 with retention of absolute
configuration. The salt 14 was prepared from the corresponding high ee secondary
benzyl alcohol.
Weinreb amides are versatile precursors to a variety of functional groups.
Stephen G. Davies of the University of Oxford devised
(Org. Lett. 2009, 11, 3254.
DOI: 10.1021/ol901174t)
a chiral Weinreb amide equivalent 17 that could be alkylated with high de.
The minor diastereomer from the alkylation was readily separable by silica gel
chromatography. Keiji Maruoka of Kyoto University established
(Angew. Chem. Int. Ed. 2009, 48, 5014.
DOI: 10.1002/anie.200901977)
that a chiral phase transfer catalyst was effective for the
enantioselective alkylation of the alkynyl ester 19.
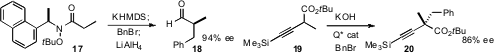
Emmanuel Riguet of the Université de Reims Champagne-Ardenne developed
(Tetrahedron Lett. 2009, 50, 4283.
DOI: 10.1016/j.tetlet.2009.05.011)
an improved catalyst for the enantioselective addition of
malonate 22 to
cyclohexenone 21. Diego A. Alonso and Carmen Nájera of the
Universidad de Alicante devised
(J. Org. Chem. 2009, 74, 6163.
DOI: 10.1021/jo9010552)
an organocatalyst for the addition of methyl acetoacetate
25 to a nitroalkene 24. José Luis García
Ruano and José Alemán of the Universidad Autónoma de Madrid observed
(Chem. Commun. 2009, 4435.
DOI: 10.1039/b908963b)
that an organocatalyst could also direct the
enantioselective conjugate addition of the bis sulfone 28 to an unsaturated
aldehyde 27. As the sulfone can be completely desulfurized, either before or
after alkylation, the net transformation is the conjugate addition of an alkyl
group.
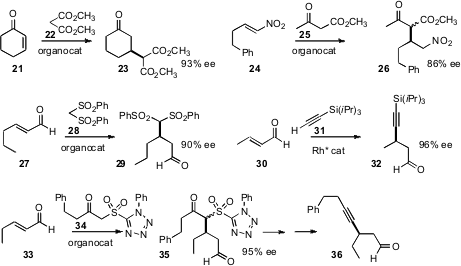
Takahiro Nishimura and Tamio Hayashi, also of Kyoto University, reported
(Angew. Chem. Int. Ed. 2009, 48, 8057.
DOI: 10.1002/anie.200904486)
a chiral Rh catalyst for the enantioselective conjugate addition of the TIPS-acetylene
31 to an unsaturated aldehyde. Karl Anker Jørgensen of Aarhus University described
(J. Am. Chem. Soc. 2009, 131, 10581.
DOI: 10.1021/ja903920j)
a complementary approach, the enantioselective addition of a sulfonyl
ketone 34. The adduct 35 could be processed into the alkyne 36, or into the
alkene (not shown).