Although there have been many synthetic approaches to morphine and its methyl
ether codeine (3), the pentacyclic structure of these Papaver alkaloids
continues to intrigue organic chemists. Philip Magnus of the University of Texas devised
(J. PMID:23659187 Am. Chem. Methyl 4-chloro-3-oxobutanoate uses Soc. 2009, 131, 16045.
DOI: 10.1021/ja9085534) an elegant
route to 3 based on the conversion of 1 to 2 by way of an intramolecular
Michael
addition. 1781098-86-9 site
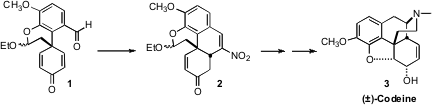
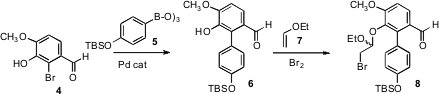
The starting point for the synthesis was the commercial bromoaldehyde 4.
Coupling with 5 delivered the substituted biphenyl 6, that was carried on to the
mixed bromo acetal 8. On exposure to fluoride ion, 8 was desilylated, and the
intermediate phenoxide cyclized with impressive facility to give 1. Exposure of
1 to nitromethane delivered the tetracyclic 2. This reaction apparently was
initiated by Henry addition of the nitromethane to the aldehyde. The
intramolecular Michael addition of the intermediate Henry adduct then proceeded
to give the desired cis diastereomer of the newly formed ring. Finally, loss of
water gave 2.
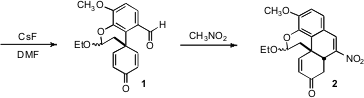
Conjugate reduction of the nitroalkene 2 led to 9 with remarkable
diastereocontrol. Exposure of 9 to
LiAlH4 converted the nitro group to the amine,
and the enone to the allylic alcohol. On exposure to acid, the hemiacetal was
hydrolyzed. The liberated aldehyde underwent reductive amination with the free
amine, while at the same time ionic cyclization closed the ether ring.
N-Acylation completed the conversion to 10.
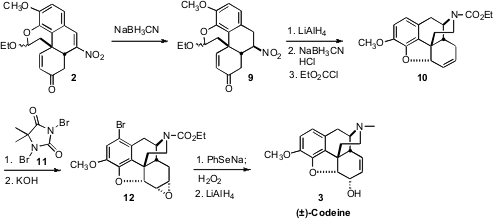
The ether 10 had previously been converted to codeine, and then, in a single
demethylation step, to morphine. In that synthesis, the alkene of 10 was directly
epoxidized. The resulting “up” epoxide reacted only sluggishly with
phenylselenide anion, and the relative configuration of the resulting allylic
alcohol had to be inverted by oxidation followed by reduction. In the current
synthesis, exposure of the alkene 10 to dibromohydantoin under aqueous
conditions, to form the bromohydrin, effected concomitant arene bromination, to
give, after base treatment, the “down” epoxide 12. Phenylselenide opening of the
epoxide was then facile, and the product allylic alcohol had the correct
relative configuration for codeine and morphine. The extra Br was of no
consequence, as it was removed by the final LiAlH4 reduction.
Except for the stereogenic center of the acetal, the dienone 1 is prochiral.
This raises the exciting possibility that it may be possible to set the absolute
configuration of codeine and thus of morphine by asymmetric catalysis of the
Henry reaction of 1.