C (Snider)
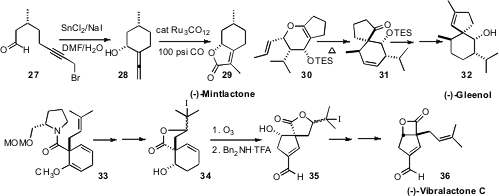
Nigel S. Simpkins, now at the University of Birmingham, found
(Chem. Commun. 2008, 5390.
DOI: 10.1039/b810441g)
that the prochiral
cyclopropane amide 1
could be deprotonated to give, after alkylation, the substituted cyclopropane
3 with high enantio- and diastereocontrol. In the course of a synthesis of
(+)-Lineatin, Ramon Alibés of the Universitat Autònoma de Barcelona optimized
(J. Org. Chem. 2008, 73, 5944.
DOI: 10.1021/jo800970u)
the photochemical cycloaddition of 4 and 5 to give, after reductive
dechlorination, the
cyclobutene 6.
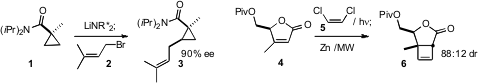
In a related reaction, José L. García Ruano and M. Rosario Martín of the
Universidad Autónoma de Madrid observed
(J. Org. Chem. 2008, 73, 9366.
DOI: 10.1021/jo801896a)
that the cycloaddition of 8 to 7 proceeded with high regio-
and diastereocontrol, to give the
cyclopentene 9. Joseph M. Ready of UT
Southwestern in Dallas developed
(Angew. Buy2166539-35-9 Chem. Int. Ed. 2008, 47, 7068.
DOI: 10.1002/anie.200801957)
a powerful new cyclopentannulation, condensing the cyclopropane
derived from the addition of 11 to 10 with the protected ynolate
12 to give 13, in the presence of a modified Lewis acid catalyst.
Chun-Chen Liao of the National Tsing Hua University, Hsinchu described
(Angew. PMID:24078122 2135443-03-5 Price Chem. Int. Ed. 2008, 47, 7325.
DOI: 10.1002/anie.200802130)
the oxidative ring contraction of the o-alkoxy phenol 14 to the
cyclopentenone 15. Stéphane Quideau of the Université de Bordeaux reported
(Org. Lett. 2008, 10, 5211.
DOI: 10.1021/ol802183p)
a related ring contraction. We uncovered
(J. Org. Chem. 2008, 73, 9479.
DOI: 10.1021/jo8017704)
a simple protocol for the in situ conversion of an ω-alkenyl
ketone such as 16 to the corresponding diazo
compound, leading, via
dipolar cycloaddition, to the adduct 17.
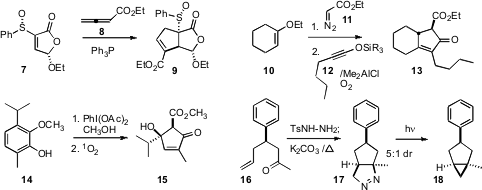
Ulrich Zutter of Roche Basel described
(J. Org. Chem. 2008, 73, 4895.
DOI: 10.1021/jo800264d)
in a synthesis of Tamiflu, the hydrogenation of 19 to give the
cyclohexane with all-cis diastereocontrol. Selective removal of the methyl ethers with
trimethylsilyl iodide set the stage for enzymatic ester hydrolysis, delivering
20 in high ee. Jonathan Clayden of the University of Manchester developed
(Angew. Chem. Int. Ed. 2008, 47, 5060.
DOI: 10.1002/anie.200801078)
a complementary approach for converting benzene
precursors to enantiomerically-pure
cyclohexenones. In a synthesis of valiolamine, Tony K. M. Shing of the Chinese University of Hong Kong carried out
(Org. Lett. 2008, 10, 4137.
DOI: 10.1021/ol801889n)
the direct aldol cyclization of 21 to 22. Catalysis
with proline gave the alternative diastereomer. Bernhard Breit of
Albert-Ludwigs-Universität, Freiburg developed
(Org. Lett. 2008, 10, 5321.
DOI: 10.1021/ol8016148)
a chiral directing group for allylic alcohol hydroformylation. Subsequent carbonyl
ene cyclization gave the
cyclohexane 24. In pursuit of the complex polycyclic
alkaloid gelsemine, Professor Simpkins reported
(Org. Lett. 2008, 10, 4747.
DOI: 10.1021/ol801835q)
a remarkable double elimination-intramolecular
Michael cyclization, converting
25 into the bridged
cyclohexanone 26.
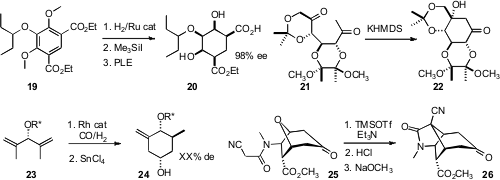
Roderick W. Bates of Nanyang Technological University found
(J. Org. Chem. 2008, 73, 8104.
DOI: 10.1021/jo801433f)
, in a synthesis of (-)-Mintlactone 29, that the diastereocontrolled
reductive cyclization of 27 to 28 worked best in wet DMF.
Susumu Kobayashi of the Tokyo University of Science showed
(Chemistry Lett. 2008, 37, 770.
DOI: 10.1246/cl.2008.770)
, en route to (-)-Gleenol (32), that the
Claisen rearrangement of
30 delivered the cyclohexene 31 with high diastereocontrol. Barry B. Snider of
Brandeis University prepared
(J. Org. Chem. 2008, 73, 8049.
DOI: 10.1021/jo8015743)
(-)-Vibralactone C (36) from 33, available from o-anisic acid by the Schultz protocol.