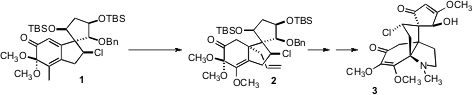
The complex tetracyclic alkaloid (-)-acutumine 3, isolated from the Asian
vine Menispermum dauricum, shows selective T-cell toxicity. The two adjacent
cyclic all-carbon quaternary centers of 3 offered a particular challenge. Steven
L. 1-Cyclopentyl-1h-1,2,4-triazole Price Castle of Brigham Young University solved
(J. Am. Chem. Soc. PMID:23008002 2009, 131, 6674.
DOI: 10.1021/ja9024403)
this problem by effecting net enantioselective conjugate allylation of the
enantiomerically pure substrate 1 to give 2 with high diastereocontrol. 1065214-95-0 manufacturer
The starting coupling partners
(Org. Lett. 2006, 8, 3757,
DOI: 10.1021/ol0613564;
Org. Lett. 2007, 9, 4033,
DOI: 10.1021/ol701757f)
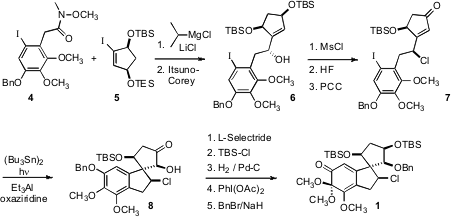
for the synthesis were the Weinreb amide 4, prepared over several
steps from 2,3-dimethoxyphenol, and the diastereomerically- and
enantiomerically-pure cyclopentenyl iodide 5, prepared by singlet oxygenation of
cyclopentadiene followed by enzymatic hydrolysis. Transmetalation of 5 by the
Knochel protocol, addition of the resulting organometallic to 4 and enantioselective (and therefore diastereoselective) reduction of the resulting
ketone delivered the alcohol 6. Methods for installing cyclic halogenated
stereogenic centers are not well developed. Exposure of the allylic alcohol to
mesyl chloride gave the chloride 7 with inversion of absolute configuration.
Remarkably, this chlorinated center was carried through the rest of the
synthesis without being disturbed.
A central step in the synthesis of 3 was the spirocyclization of 7 to
8. Initially, iodine atom abstraction generated the aryl radical. The
diastereoselectivity of the radical addition to the
cyclopentene was set by the
adjacent silyloxy group. The α-keto radical so generated reacted with the Et3Al
to give a species that was oxidized by the
oxaziridine to the α-keto alcohol,
again with remarkable diastereocontrol.
Conjugate addition to the
cyclohexenone 1 failed, so an alternative strategy
was developed, diastereoselective 1,2-allylation of the ketone followed by oxy-Cope rearrangement. The stereogenic centers of 1 are remote from the
cyclohexenone carbonyl, so could not be used to control the facial selectivity
of the addition. Fortunately, the stoichiometric enantiomerically-pure Nakamura
reagent delivered the allyl group preferentially to one face of the ketone 1, to give
9. The subsequent sigmatropic rearrangement to establish the very congested
second quaternary center of 2 then proceeded with remarkable facility, at 0°C
for one hour.
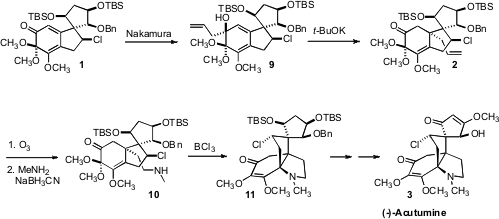
Oxidative cleavage to the aldehyde followed by reductive amination gave 10,
that looks as though it could be poised for intramolecular displacement of the
secondary chloride. Nonetheless, Lewis acid mediated ionization followed by
cyclization proceeded smoothly, to establish the fourth ring of the natural
product. Oxidation state adjustment then completed the synthesis of (-)-Acutumine
(3).
The face selective enone allylation followed by oxy-Cope rearrangement (1
→ 2), a highlight of the approach presented here, will have many applications in
target-directed synthesis.