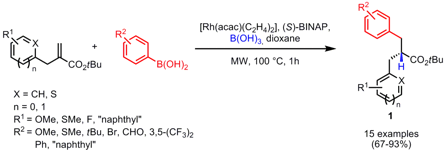
The asymmetric synthesis of α,α’-dibenzyl esters 1 from α-benzyl acrylates and diversely substituted boronic acids was performed by Christopher Frost and co-workers from University of Bath (Org. 1345728-51-9 Formula Lett. 2007, 9, ASAP.DOI: 10.1021/ol070603g). Price of 152835-00-2 Dibenzyl esters 1 were generally obtained in good enantioselectivities via a tandem rhodium-catalyzed conjugate addition-enantioselective protonation protocol. (S)-BINAP as ligand and B(OH)3 as proton source proved to be the best combination in terms of yields and enantioselectivities in optimization studies.
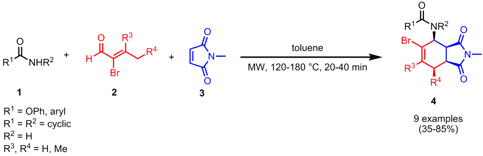
Three-Component Synthesis of 1-Amido-2-cyclohexenes
The group of Matthias Beller from the University of Rostock has reported on the three-component reaction of amides 1, aldehydes 2 and dienophiles3 (AAD reaction) to give 2-bromo-substituted 1-amido-2-cyclohexene derivatives 4 (Synlett2007, 1085. PMID:23903683 DOI: 10.1055/s-2007-973898). With this protocol aromatic amides, sulfonamides, carbamates and cyclic amides are well tolerated, the latter providing the products only in moderate yields (35-46%). Up to four stereogenic centers are generated in the AAD sequence and in most cases an all-syn-configuration is obtained. By applying α-bromo aldehydes 2, further scaffold decorations like cross-coupling or carbonylation reactions are possible at the 2-position.
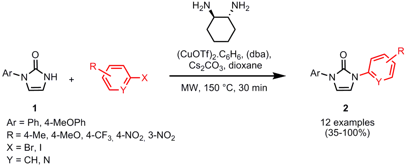
Synthesis of Unsymmetrically Substituted N,N’-Diarylimidazolin-2-ones
A set of unsymmetrically substituted N,N’-diarylimidazolinones 2 was synthesized by Doris Kunz and Thomas Hafner from Ruprecht-Karls-University Heidelberg via copper-catalyzed N-arylation of arylimidazolinones 1 with aryl iodides or bromides (Synthesis 2007, 1403. DOI: 10.1055/s-2007-966021). Similar yields can be achieved by conventional heating at lower temperature (120 °C), however, a longer reaction time of 15 h has to be applied. The synthesis of symmetrically substituted N,N’-diarylimidazolinones via double N-arylation was less successful since substrate decomposition of imidazolinone occurred as a side reaction. Thus only electron-poor aryl halides can be employed which furnish the products in moderate yield.
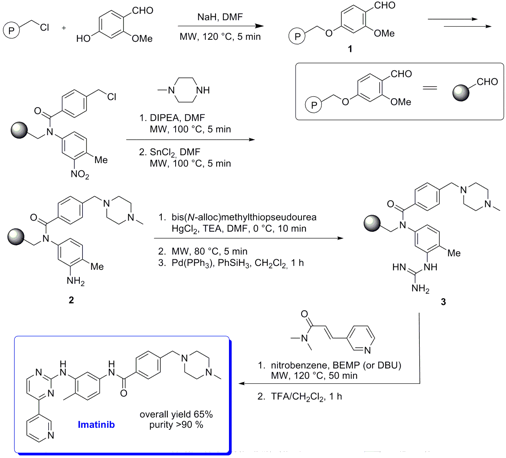
Solid-Phase Synthesis of Imatinib
Angelo Carotti and his group from University of Bari have developed a solid-phase synthesis of Imatinib which acts as a selective tyrosine kinase inhibitor (Tetrahedron Lett. 2007, 48, 3455. DOI: 10.1016/j.tetlet.2007.03.033). By applying microwave heating in five steps of the synthesis (preparation of linker 1, nucleophilic substitution, reduction of the nitro group, formation of guanidine and final cyclization) the total process could be accelerated. Key steps were the guanylation of aniline 2 where a higher yield and purity of product 3 could be obtained under microwave irradiation, and the final cyclization to resin bound Imatinib where the reaction time could be reduced from 20 h to 50 min. Addiotionally, resin stability was ensured due to the shorter reaction time.