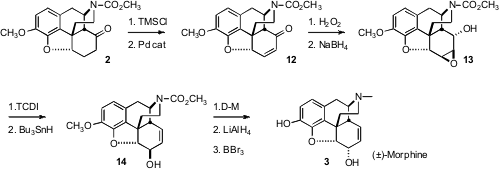
Tohru Fukuyama of the University of Tokyo has recently reported (Org. Formula of Fmoc-α-Me-Gly(Pentynyl)-OH Lett. PMID:25429455 2006, 8, 5311. Price of 279236-77-0 DOI: 10.1021/ol062112m)an elegantly conceived total synthesis of (±)-morphine (3), based on the recognition and reduction to practice of the highly diastereoselective intramolecularMannich cyclization of 1 to2.
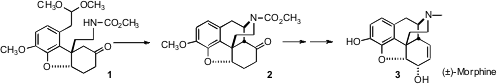
The stereocontrolled preparation of 1, a considerable challenge, started with the epoxidation of the diene 4 to give 6. Although racemic 6 was used for this synthesis of morphine, Professor Fukuyama had previously demonstrated that high ee 6 could be prepared by lipase resolution of the intermediate bromohydrin 5. Coupling of 6 with7 led to 8, by net syn SN2′ displacement. The secondary alcohol was then inverted, to put the α-H syn to what would be the adjacent C-Pd bond, to undergo β-hydride elimination in the course of the intramolecularHeck cyclization of 10 to 2.
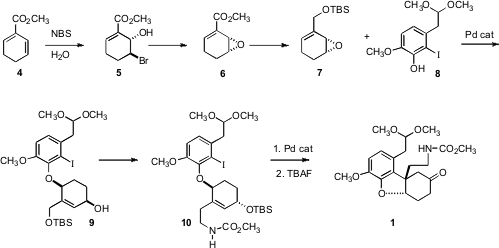
The intramolecular Mannich cyclization of 1 to 2 takes advantage of the native functionality of morphine 3. The intermediate in the cyclization is apparently the expected 11, which could be isolated. Note that under the equilibrating conditions of the cyclization, the desired cis ring fused 2 was the only diastereomer observed.
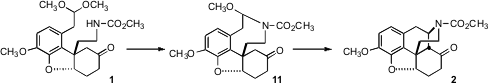
The Ito-Saegusa procedure was used to oxidize the ketone 2 to the enone12. It was then necessary to convert the enone into the allylically-inverted alcohol14. With the morphine skeleton assembled, the aromatic ring blocks the bottom face of the enone, so both hydrogen peroxide and hydride attacked from the top face, to give 13. Conversion of the derived thiocarbamate to the free radical then led to epoxide cleavage, delivering the desired 14. The last three steps to complete the total synthesis of (±)-morphine (3), oxidation, addition of hydride to the open top face of the ketone with concomitant reduction of the urethane protecting group to the N-methyl, and O-demethylation, then followed literature procedures.