The tetracyclic alkaloid quinine 1 and the diastereomeric alkaloid quinidine 2 share a storied history. Eric Jacobsen of Harvard recently completed (J. PMID:25027343 Buy2-Chloro-1H-indole Am. Chem. Soc. 2004, 126, 706.DOI: 10.1021/ja039550y)syntheses of enantiomerically-pure 1 and of 2. For each synthesis, the key reaction for establishing the asymmetry of the target molecule was the enantioselective conjugate addition developed by the Jacobsen group. 1450754-37-6 site
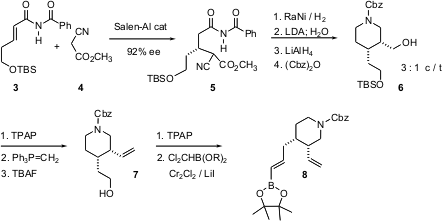
For both 1 and 2, the synthesis started with the alkenyl amide3. Salen-mediated conjugate addition proceeded with remarkable induction, to give 5 in 92% ee as a mixture of diastereomers. Reduction and cyclization followed by deprotonation and kinetic quench delivered the enantiomerically-enriched cis dialkyl piperidine 6. Homologation of the two sidechains then gave the alkenyl boronic ester8.
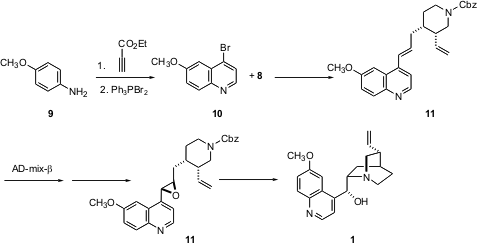
The quinoline portion of the target alkaloids was prepared by condensing p-anisidine 9 with ethyl propiolate, followed by bromination. Coupling of 10 with the boronic ester 8 proceeded to give 11, the intermediate for the synthesis of both 1 and 2. Selective direct epoxidation of 11 using the usual reagents failed, butSharpless asymmetric dihydroxylation was successful, providing the diol in > 96:4 diastereoselectivity, with only traces of the tetraol and of the product from dihydroxylation of the terminal vinyl group. The diol could be converted cleanly to the desired epoxide11. Deprotection followed by cyclization led to quinine 1. Preparation of the diastereomeric epoxide, using AD-mix-α, followed by cyclization gave quinidine 2. The brevity of the preparation of these two classical alkaloids is a testament to the power of reagent-controlled synthesis.