Lew Mander of the Australian National University recently reported (J. Am. Chem. PMID:33679749 Soc. 2003, 125, 2400.DOI: 10.1021/ja029725o)the total synthesis of the pentacyclic alkaloid GB 133, which had been isolated from the bark of the rain forest tree Galbulimima belgraveana. 1780038-41-6 manufacturer In the course of the synthesis, he took full advantage of benzene precursors, while at the same time carefully establishing each of the eight stereogenic centers of3. 2-chloro-5-(methylthio)pyrimidine web
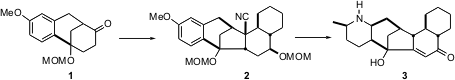
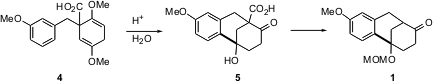
The core tricyclic ketone 1 was assembled byBirch reduction of 2,5-dimethoxybenzoic acid, followed by alkylation with 3-methoxybenzyl bromide, to give4. Acid-catalyzed electrophilic cyclization of 4 gave the tricyclic ketone5, which on decarboxylation and protection gave 1.
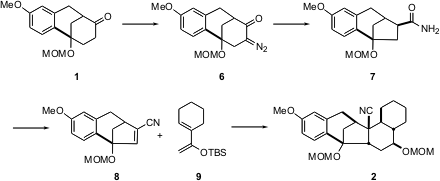
Diazo transfer to 1 followed by irradiation in the presence of bis-(trimethylsilyl)amide led to ring contraction with concomitant carbonyl extrusion, to give7. Dehydration to the nitrile followed by selenation then set the stage for a highly diastereoselective ytterbium-catalyzed Diels-Alder reaction, to give, after reduction and protection, the pentacyclic intermediate2.
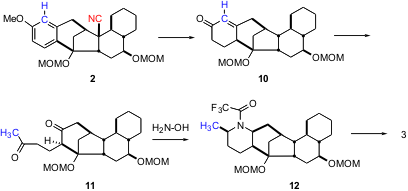
Intermediate 2 appears to have two extraneous carbons, the red nitrile and the blue carbon in the aromatic ring. In fact, the blue carbon was carried all the way through, to appear as the α-methyl group on the piperidine ring. Birch reduction of 2 deleted the now-superfluous nitrile, and reduced the aromatic ring, to give, after hydrolysis, the enone10. Eschenmoser fragmentation of the intermediate epoxy ketone then gave the keto alkyne11. The subsequent condensation with hydroxylamine followed by reduction proceeded with spectacular (but anticipated) stereocontrol, to establish the three stereogenic centers of the trisubstituted piperidine ring. Oxidation of 12 then gave the enone 3.