(Opatz), (-)-Lycoperine A (Rychnovsky), Fluvirucidine A2 (Suh), Complanidine A (Sarpong)
Günter Helmchen of the Ruprecht-Karls-Universität Heidelberg set
(Org. 1450752-97-2 In stock Lett. 2010, 12, 1108.
DOI: 10.1021/ol1001076)
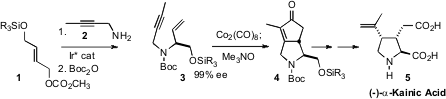
the absolute configuration of 3
by Ir*-mediated coupling of 1 with 2. Diastereoselective
Pauson-Khand cyclization then led to (-)-α-Kainic Acid (5).
Till Opatz, now at the Johannes Gutenberg-Universität Mainz, showed
(Org. 1226800-12-9 site Lett. 2010, 12, 2140.
DOI: 10.1021/ol100652b)
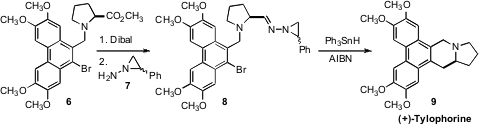
that the product from the
Dibal reduction of 6 could be
condensed with the amine 7 without epimerization. PMID:25016614 Kim cyclization then
directly delivered the pentacyclic alkaloid (+)-Tylophorine (9).
The interesting dimeric alkaloid Lycoperine A (13) was recently
isolated from the Japanese club moss Lycopodium hamiltonii. Scott D.
Rychnovsky of the University of California, Irvine prepared
(Org. Lett. 2010, 12, 72.
DOI: 10.1021/ol902389e)
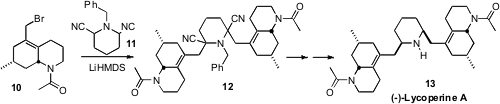
12 by double alkylation of the bis-nitrile
11 with the enantiomerically-pure allylic bromide 10. While the
projected reductive decyanation of 12 failed, hydrolysis followed by
diastereoselective reductive amination successfully gave 13.
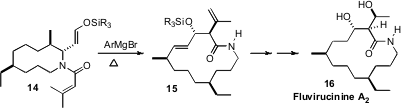
Retrosynthetic analysis of Fluvirucinine A2 (16) could lead
to an acyclic amino acid, that could be cyclized to the macrolactam. Young-Ger
Suh of Seoul National University took
(Org. Lett. 2010, 12, 2040.
DOI: 10.1021/ol100521v)
a different approach, building up the fourteen-membered ring system by two
four-carbon ring expansions, beginning with an enantiomerically-pure piperidine
precursor. The second of these enolate-based aza-Claisen ring expansions is
illustrated in the conversion of 14 to 15.
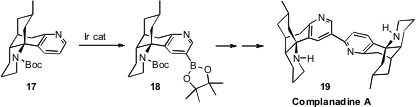
Richmond Sarpong of the University of California, Berkeley faced
(J. Am. Chem. Soc. 2010, 132, 5926.
DOI: 10.1021/ja101893b)
a different sort of challenge in the synthesis of the dimeric
Lycopodium alkaloid Complanadine A (19).
Even with established access to monomers such as 17 and its precursors,
it was not clear how the 5-position of the pyridine ring could be selectively
activated for bond formation. The solution to this dilemma was found in the work
of Hartwig. Following that precedent, Ir-catalyzed activation of 17
converted it cleanly into the boronate 18, that could then be coupled
with a pyridone triflate to complete the synthesis of 19.