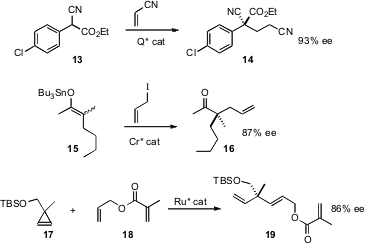
Oxygenated secondary stereogenic centers are readily available. There is a
limited range of carbon nucleophiles that will displace a secondary leaving
group in high yield with clean inversion. Teruaki Mukaiyama of the Kitasato
Institute has described (Chem. Lett. 2007, 36, 2.
DOI: 10.1246/cl.2007.2)
an elegant addition to this
list. Phosphinites such as 1 are easily prepared from the corresponding
alcohols. Quinone oxidation in the presence of a nucleophile led via efficient
displacement to the coupled product 2. 1934533-59-1 Purity The sulfone could be
reduced with SmI2 to give
3.
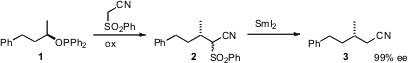
Enantioselective reduction of trisubstituted alkenes is also a powerful
method for establishing alkylated stereogenic centers. Juan C. Carretero of the
Universidad Autonoma de Madrid has found (Angew. Chem. Int. Ed. 4,7-Dibromo-1H-1,3-benzodiazole Chemscene 2007,
46, 3329.
DOI: 10.1002/anie.200700296)
that the enantioselective reduction of unsaturated pyridyl sulfones such as
4
was directed by the sulfone, so the other geometric isomer of 4 gave the
opposite enantiomer of 5. The protected hydroxy sulfone 5 is a versatile chiral
building block.
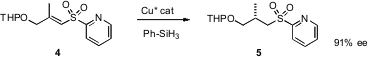
Samuel H. Gellman of the University of Wisconsin has reported (J. Am. Chem. Soc. PMID:23907051
2007, 129, 6050.
DOI: 10.1021/ja070063i)
an improved procedure for the aminomethylation of
aldehydes. L-Proline-catalyzed condensation with the matched α-methyl
benzylamine derivavative 7 gave the aldehyde, which was immediately reduced to
the alcohol 8 to avoid racemization. The amino alcohol 8 was easily separated in
diastereomerically-pure form.
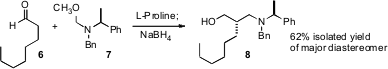
In the past, aldehydes have been efficiently α-alkylated using two-electron
chemistry. David W. C. Macmillan of Princeton University has developed (Science
2007, 316, 582,
DOI: 10.1126/science.%201142696;
J. Am. Chem. Soc. 2007, 129, 7004,
DOI: 10.1021/ja0719428)
a one-electron alternative.
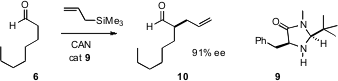
The organocatalyst 9 formed an imine with the aldehyde. One-electron oxidation
led to an α-radical, which was trapped by the allyl silane (or, not pictured, a
silyl enol ether) leading to the α-alkylated aldehyde 10. This is mechnistically
related to the work reported independently by Mukund P. Sibi (J. Am. Chem. Soc.
2007, 129, 4124,
DOI: 10.1021/ja069245n;
![]()
2008, February 11) on one-electron α-oxygenation of aldehydes.
Secondary alkylated centers can also be prepared by SN2′ alkylation of prochiral substrates such as
11. Ben L. Feringa of the University of Groningen
has shown (J. Org. Chem. 2007, 72, 2558.
DOI: 10.1021/jo0625655)
that the displacement proceeded with
high ee even with conventional Grignard reagents. The products so formed are
versatile intermediates for further transformation.
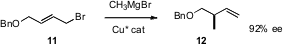
The enantioselective construction of acyclic alkylated quaternary stereogenic
centers is a continuing challenge in organic synthesis. Several promising
approaches have recently appeared. Li Deng of Brandeis University has
established conditions (J. Am. Chem. Soc. 2007, 129, 768.
DOI: 10.1021/ja0670409)
for the catalyzed
conjugate addition of aryl cyanoacetates such as 13 to acrylonitrile to give the
adduct 14 in high ee. Eric N. Jacobsen of Harvard University has developed (Angew.
Chem. Int. Ed. 2007, 46, 3701.
DOI: 10.1002/anie.200604901)
a chiral Cr catalyst that mediated the alkylation
of tributyltin enolates such as 15 to give 16 in high ee. Amir H. Hoveyda of
Boston College has designed (J. Am. Chem. Soc. 2007, 129, 3824.
DOI: 10.1021/ja070187v)
a chiral Ru
metathesis catalyst that crossed 18 with the readily-prepared prochiral
17 to give 19, also in high ee.