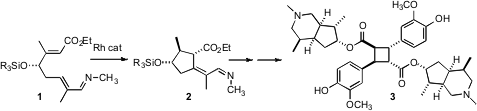
The monoterpene alkaloid (-)-incarvillateine (3) has interesting symmetry properties. The centralcyclobutane diacid core is not itself chiral, but the appended alkaloids are. The key step in the total synthesis of 3 recently (J. Am. Chem. PMID:23724934 Soc. Bis(pyridine)iodonium tetrafluoroborate Data Sheet 2008, 130, 6316.DOI: 10.1021/ja8012159)described by Robert G. Bergman and Jonathan A. Methyl 4-ethynylbenzoate site Ellman of the University of California, Berkeley was the diastereoselective Rh-catalyzed cyclization of 1 to 2.
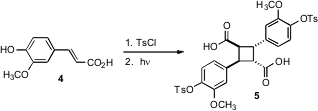
The cyclobutane diacid core 5 was assembled from ferulic acid 4 following the procedure of Kibayashi (J. Am. Chem. Soc. 2004, 126, 16553.DOI: 10.1021/ja0401702).
The starting point for the preparation of 1 was the commercial aldehyde6. Enantioselective allylation followed by silylation delivered 7, which on cross metathesis with methacrolein gave the diene aldehyde 8. Imine formation then completed the construction of 1.
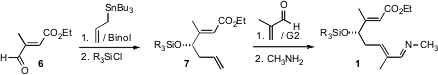
The cyclization of 1 was effected by warming (45°C, 6 h) with 2.5 mol % [RhCl(coe)2]2 and 5.5 mol % (DMAPh)Pet2 ligand. While four diastereomers were possible from the cyclization, only two were observed, with one predominating. Since the product mixture was easily susceptible to tautomerization, it was carried on directly to reduction and cyclization, to form the lactam 8.
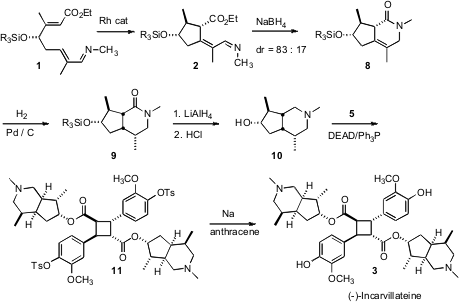
Hydrogenation of 8 to 9 required high temperature and pressure, but delivered9 as a single diastereomer. Reduction and desilylation then set the stage forMitsunobu coupling with 5, to give 11. Dissolving metal conditions removed the tosyl groups from 5 to give (-)-incarvillateine 3.
It will be interesting to see how general this Rh catalyzed cyclization will be. It will also be interesting to establish the mechanism. The authors described the cyclization of 1 as proceeding via initial metalation of the alkene C-H bond, followed by insertion of the ester-bearing alkene into the C-Rh bond to form a new C-Rh bond, and finally reductive elimination. Their previous observation of metalation of such an unsaturated imine with maintenance of the alkene geometry suppported this mechanism. The high diastereocontrol also suggested intramolecular C-C bond formation. Whatever the mechanism, the enantiomerically-pure cyclopentane 2, having four of its five carbons functionalized, is a versatile intermediate for further transformation.