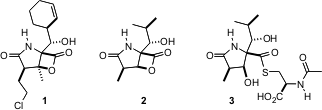
Inhibitors Salinosporamide A, Omuralide and Lactacystin
The structurally-related
γ-lactams salinosporamide A (1), omuralide (2)
and lactacystin (3), of bacterial origin, inhibit proteasome activity,
and so are of interest as lead compounds for the development of anticancer
agents. Barbara C. M. Potts of Nereus Pharmaceuticals in San Diego has reported
(J. Med. Chem.
2005, 48, 3684.
DOI: 10.1021/jm048995+)
a
detailed structure-activity studies in this series, and E.J. Corey of Harvard
University has prepared (J. 1350518-27-2 web Am. PMID:23310954 1206981-68-1 Formula Chem. Soc. 2005,
127, 8974,
DOI: 10.1021/ja052376o; 15386,
DOI: 10.1021/ja056284a.)
several interesting structural analogues. Susumi Hatakeyama of
Nagasaki University, building on previous work in this area, has reported (J.
Org. Chem.
2004, 69, 7765.
DOI: 10.1021/jo048817o)
a
synthesis of 2 and 3 from Tris.
In cell culture, 1 is by far the most active of these three natural
products. The challenge in the synthesis of 1 is not closing the
β-lactone, but rather the stereocontrolled assembly of the γ-lactam 9.
E.J. Corey reported (J. Am. Chem. Soc.
2004, 126, 6230.
DOI: 10.1021/ja048613p)
the first
route to
1. The acrylamide 5 was prepared from (S)-threonine methyl
ester. Highly diastereoselective (9:1) intramolecular
Baylis-Hillman
condensation of 5 followed by silyation led to 6, which cyclized
under Stork conditions to the cis-fused 7. Addition of
cyclohexenyl zinc proceeded with remarkable diastereocontrol, to give 8.

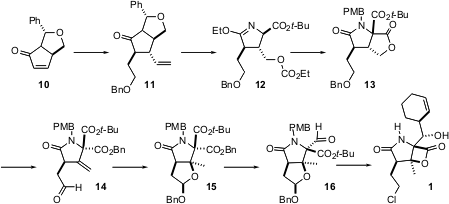
Samuel Danishefsky of Columbia University has also described (J. Am. Chem.
Soc.
2005, 127, 8298.
DOI: 10.1021/ja0522783)
a total
synthesis of
1, starting from the pyroglutamate derivative 10. Conjugate
addition followed by alkylation established the lactam framework. Intramolecular
cyclization of 12 gave 13, establishing the aminated quaternary
center. The oxygenated quaternary center was then constructed by
phenylselenyl-mediated cyclization of 14. The end game of this synthesis
used the already-established cyclohexenyl zinc addition, which worked as well
with 16 as it had with 7.
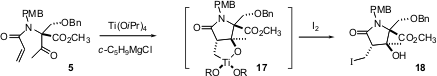
E. J. Corey recently described (Org. Lett. 2005, 7,
2699,
DOI: 10.1021/ol0508734; 2703,
DOI: 10.1021/ol050874w.) an even
more elegant approach to the γ-lactam. Exposure of 5 to the
Kulinkovich
conditions followed by iodination delivered 18, presumably by way of the
titanacycle
17. Although all-carbon trans-5,5 systems are strained, the
trans ring fusion is the expected stereochemical outcome with (RO)2Ti
in the ring. Complexation of the ester O to the Ti may explain the observed
facial selectivity. Brief exposure of
18 to Et3N followed by silylation converted it to 6.
The preparation of several more analogues in this series is also reported in
these two articles.