The aldol addition reaction is one of the most versatile, effective and general methods for the formation of C-C bonds in modern organic synthesis. The challenge raised by nature has stimulated many scientists who, over the last decades, have established the aldol reaction as the main tool to access important synthons for constructing the polyol architecture that is present in many natural products. This highlight focuses on developments concerning the cross-aldol reactions of aldehydes.
The classic aldol addition faces the problems of chemo- and regioselectivity as well as that of asymmetry. The 2001 Nobel Prize awarded to K. Barry Sharpless, Ryoji Noyori and William S. Knowles, for their research in the use of chiral auxiliaries, ligands and catalysts for stereochemical control, outlines the importance of this topic. However, the aldol reaction of aldehydes has some drawbacks, and a general solution to the problem of stereoselective control has not yet found.

Scheme 1 – Problems associated with metal-catalysed aldol reactions of aldehydes (PNAS 2004, 101, 5439. DOI: 10.1073/pnas.0307212101).
Several problems are associated with aldol reactions of aldehydes due to the fact they suffer from undesired reactions, such as polyaldolisation (secondary additions to the aldehyde products), oligomerisation of the product (propensity of aldehydes to polymerise under metal-catalysed conditions), dehydration of the product and Tishchenko-type processes (Scheme 1).
Recently, the catalytic asymmetric direct cross-aldol reactions of aldehydes have been reviewed by Alcaide and Almendros (Angew. Chem. Int. Ed. 2003, 42, 858. DOI: 10.1002/anie.200390232).
The present highlight is divided in three main sections: the cross-aldol of aldehyde enolates with aldehydes, catalytic methods for the direct cross-aldol, and the reductive cross-aldol reaction.
II. The Cross-Aldol reaction of aldehyde enolates with aldehydes
The work developed by the Heathcock group on theE and Z lithium enolates of propanal showed some limitations concerning low selectivity (J. Org. Chem. 1980, 45, 1066). The cross-aldol reaction between an aldehyde and an α,β-unsaturated aldehyde via titanium enolates was reported by the Oshima group (Tetrahedron Lett. Formula of 1-Bromo-4-chloro-2,5-difluorobenzene 2000, 41, 4415. DOI: 10.1016/S0040-4039(00)00642-0). The use of Ti(O-n-Bu)4/t-BuOK (1:1) substantially improved the yields of β-hydroxyaldehydes (Scheme 2). The authors outlined the fact that the molar ratio of Ti(O-n-Bu)4/t-BuOK plays an important role in the success of the reaction. Although good yields were obtained (up to 91%), stereoselectivity was low and dependent on enolate geometry.
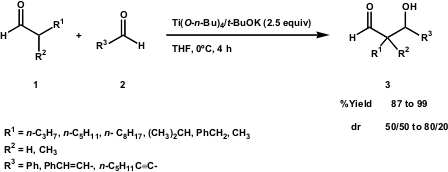
Scheme 2 – Cross-Aldol condensation of aldehydes using a combined reagent of Ti(O-n-Bu)4/t-BuOK (1:1).
Scott E. Denmark from the University of Illinois has recently developed a chiral phosphoramide-catalysed, enantioselective cross-aldol reaction of aldehydes (PNAS 2004, 101, 5439. DOI: 10.1073/pnas.0307212101)(Figure 1).
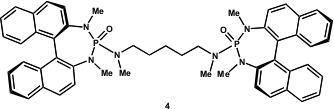
Figure 1 – Chiral bis-phosphoramide (R,R)-catalyst used by the Denmark group.
Despite the problems associated with silyl enol ethers derived from aldehydes, such as their propensity for undesired reactions, Denmark presented an alternative approach using a chiral Lewis base and reactive trichlorosilyl enolates (Angew. PMID:25027343 Chem. Int. Ed. tert-Butyl 5-aminopentanoate manufacturer 2001, 40, 4759. DOI: 10.1002/1521-3773(20011217)40:24%3C4759::AID-ANIE4759%3E3.0.CO;2-G). The unstable β-hydroxyaldehyde reaction products were isolated as dimethyl acetals.
Moreover, excellent diastereoselectivity was achieved in Denmark’s most recent approach, which depends on enolate geometry as well as with catalyst configuration.

Scheme 3 – Preparation of isobutyraldehyde trichlorosilyl enolate6 directly from aldehyde 5.
Enolate 6, prepared under mild conditions in a single step procedure (Scheme 3), underwent aldol addition to several aldehydes 7 in the presence of 10 mol% of phosphoramide (R,R)-4 (Scheme 4), and the corresponding products were protected as dimethyl acetals. The authors found no aldol product when the reaction was performed in the absence of Lewis base catalyst 4. A wide variety of substrates with distinct electronic characteristics were tested, such as aromatic, olefinic and aliphatic aldehydes. All aldehydes gave the desired product in high yields (80-92%) and moderate to good selectivity. Surprisingly, both electron-rich and electron-poor aldehydes gave increased selectivity. Another interesting feature is that aldol adducts from both electron-poor and electron-rich aldehydes gave the same product configuration (absolute configuration was established by X-ray analysis). The authors are still working on the interpretation of these observations and on mechanistic studies.
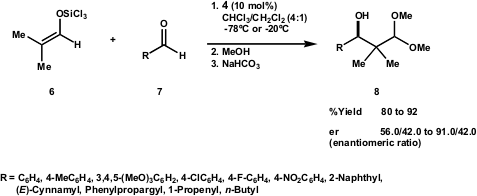
Scheme 4 – Preparation of isobutyraldehyde trichlorosilyl enolate 6.
The Denmark approach provided a simple method for the preparation of enolate6 on a multigram scale, as well as a direct procedure for preparing β-hydroxyaldehyde units in high yields with minimal functional group manipulation.
Aldol reactions catalysed by enzymes are also important to mention. DERA, or 2-deoxyribose 5-phosphate aldolase, is an enzyme that catalyses C-C bond formation, often in a highly stereoselective way, and is the only enzyme that accepts more than one aldehyde in the condensation reaction (J. Am. Chem. Soc. 1995, 117, 7585).
III. The Direct Cross-Aldol Reaction – Catalytic Methods
The Mahrwald group developed one of the first methods for the diastereoselective aldol addition of an enolisable aldehyde to another aldehyde (Tetrahedron Lett. 1997, 38, 4543. DOI: 10.1016/S0040-4039(97)00984-2). This procedure involved a Lewis-acid complex of the aldehydes, which underwent aldol addition in the presence of a base, either TMEDA (N,N,N’,N’-tetramethylethylendiamine) or TEA (triethylamine). However, only syn-selectivity was observed.
The remarkable work developed by Barbas, List, Shibasaki and Trost in enantioselective direct aldol reactions prompted the MacMillan group to investigate the enantioselective aldehyde-aldehyde coupling using a catalytic method (J. Am. Chem. Soc. 2002, 124, 6798. DOI: 10.1021/ja0262378). This group reported the first direct enantioselective catalytic cross-aldol, where the chiral amine L-proline acted as an aldolase enzyme mimic when used as catalyst. β-Hydroxyaldehydes were obtained in high yield (up to 87%) and high enantioselectivity (up to 99% ee) (Scheme 5). The main contribution of this method is that aldehydes that contain an enolisable α-methylene proton can be used as both the aldol acceptor and donor.
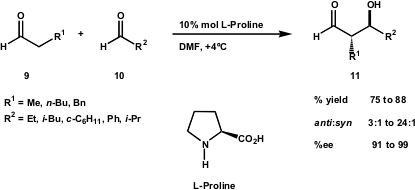
Figure 2 – 6´-epi-peridinin structure
Recently, the McMillan group reported (Tetrahedron 2004, 60, 7705. DOI: 10.1016/j.tet.2004.04.089)the use of aldehyde α-thioacetals as versatile acceptors in direct proline-catalysed aldol reactions to produce anti-aldol adducts with high diastereo- and enantiocontrol (Scheme 6). McMillan’s choice of α-thioacetals aldehydes was made on the basis of the synthetic utility of thioacetals as carbonyl and alkyl equivalents, as well as the expectation that they would be highly electrophilic, sterically and electronically deactivated towards enamine formation, and that the steric demand of the α-dithiane would improve the absolute and relative stereocontrol. The authors underscore the fact that propanal must be added slowly to an excess (2 equiv) of the electrophilic aldehyde in order to suppress dimerisation, which simultaneously increases the levels of anti-diastereoselectivity and enantiocontrol. Experiments carried out with cyclic thioacetals aldehydes gave better results. This observation was explained on the basis of a decreased reactivity of the acceptor face due to the steric demand of the acetal.
McMillan’s approach allowed access with high levels of diastereocontrol to cross-aldol adducts that are very important to biological polyol structures, without pre-generation of enolate equivalents and with the advantage that aldehyde α-thioacetals can be prepared on large scale and stored.
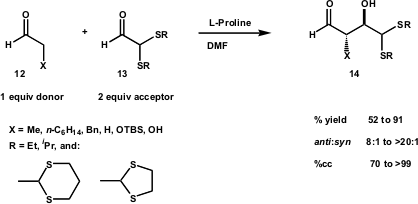
Scheme 6 – Enantioselective direct cross-aldol reaction using aldehyde α-thioacetals.
IV. The Reductive Cross-Aldol Reaction
A reductive cross-aldol reaction was recently developed by the Baba group (Org. Lett. 2005, 7, 1845. DOI: 10.1021/ol050533i), employing an α-bromoaldehyde, an aldehyde and Ge(II) as a reductant (Scheme 7). The syn-cross-aldol products 17 were isolated in moderate to high yields.
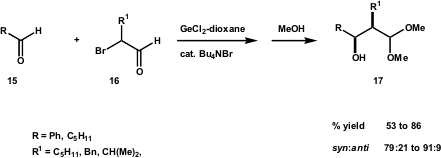
Scheme 7 – Reductive cross-aldol reaction between different aldehydes mediated by germanium(II).
According to the authors, the use of GeCl2 avoided the over-reaction observed when SnCl2 was used instead. The authors draw attention to the order of addition, which proved to be very important. The bromoaldehyde should be added last, since lower yields were obtained when it was premixed with GeCl2. The addition of Bu4NBr as an additive increased the yields dramatically, and in some cases a higher amount of this salt increased the diastereoselectivity as well. The Baba group performed an intramolecular reaction using this procedure. The use of Germanium (II) to generate the aldehyde enolate under mild, non-basic conditions via metal-halogen exchange is related to the Reformatsky reactions of bromoesters.
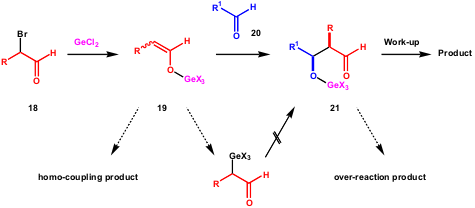
Scheme 8 – Plausible reaction course and pathways for undesired side-reactions proposed by the Baba group.
A one pot synthesis was also realised, where a bromination followed by a reductive cross-aldol reaction afforded the desired adduct in 65% yield and a syn:anti ratio of 83:17.
The Baba group proposed a plausible reaction course (Scheme 8). Once the germanium enolate 19 forms, it adds to aldehyde 20 resulting in 21. The effect of Bu4NBr is explained as playing the role of a ligand to the germanium centre, which increases the nucleophilicity of 19. The major product is the syn-adduct, which they propose is formed via an acyclic transition state. Scheme 8 also shows the undesired reactions that might occur. It is important to emphasise that germanium oxide 21 does not participate in further nucleophilic additions.
V. Summary
The cross-aldol reaction of aldehydes constitutes a formidable synthetic challenge for chemists. Over the last five years, several groups have made strong contributions to the development of efficient and elegant procedures that permit access to important synthons in polyol natural products. Some of the more impressive achievements have been reviewed in this highlight.