Carotenoids are a class of natural pigments found mainly in algae, plants and some bacteria. From the several roles of carotenoids, the biological activity as antioxidants and sources of vitamin A (1) (Figure 1) are amongst the most important. Retinoids are a group of natural and synthetic analogues of retinal [vitamin A (1)], and their most relevant activity is during the development of the embryo and in postnatal life. Structural features such as the nature of the end group and geometry of the polyene side chain are extremely important for the biological activity of retinoids.
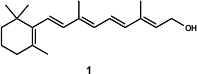
Figure 1 – Vitamin A structure
Recently, extensive efforts have been devoted to the stereocontrolled total synthesis of carotenoids and retinoids. The strategies developed for the polyenic chain construction were based on the Wittig, Horner-Wadsworth-Emmons, Julia and related reactions. However, mixtures of E/Z isomers were produced and difficult to separate. This review focuses on the newest approaches developed over the past 4 years where this problem was successfully overcome using a Stille reaction as the key step.
II. Price of 1131912-76-9 Synthesis of Carotenoids and Retinoids
The Negishi and Zeng approach (Org. Lett. 2001, 3, 719.DOI: 10.1021/ol000384y)to carotenoids and related natural products was based on a Pd- and Zn-catalyzed alkenyl-alkenyl coupling (Scheme 1). The synthesis of β-carotene (6) was performed via tetraenyne 5. The authors used a novel methodology which consisted of an Al-Zn transmetallation of alkynes, and the resulting alkenylalanes were coupled with the use of 3 and 4 as synthons, in a cross-coupling reaction catalysed by Pd and Zn. This route proved to be efficient and stereoselective (> 98% isomeric purity) not only for the synthesis of symmetrical as well as unsymmetrical carotenoids, but also for retinoids.
Scheme 1 – The Negishi and Zeng synthesis of β-carotene (6), with 53% yield and > 98% isomeric purity.
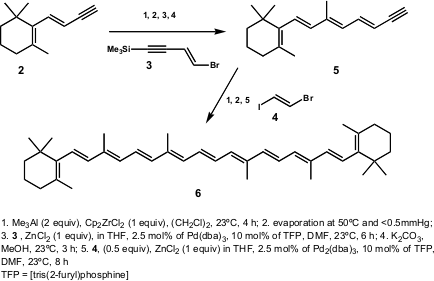
Katsumura and co-workers (Angew. Chem. Int. PMID:23522542 Ed. 2002, 41, 1023.DOI: 10.1002/1521-3773(20020315)41:6%3C1023::AID-ANIE1023%3E3.0.CO;2-A)reported the total synthesis of the carotenoid Peridinin (7). The retrosynthetic approach developed by this group is shown in Scheme 2. 53103-03-0 Chemscene
Scheme 2 – The retrosynthetic approach developed by Katsumura and co-workers for the synthesis of Peridinin (7).
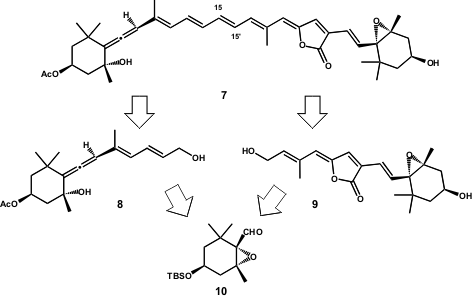
The strategy envisaged the division of 7 in two main fragments 8and 9 (central double bond C15-C15´), the coupling which constituted the last step of the synthesis. The authors performed the coupling of 8 and9 with a modifiedJulia-Kocienski olefination (Scheme 3). Thus, the allene 8 was converted into sulfone 11 and coupled with aldehyde12 (prepared from 9), at -78ºC in the dark. The authors outlined the fact that, although the crude product was a mixture of E– and Z-isomers (1:3), the Z-isomer could be slowly converted into E-isomer (7) in benzene solution over 3 days in the dark (E/Z > 5:1).
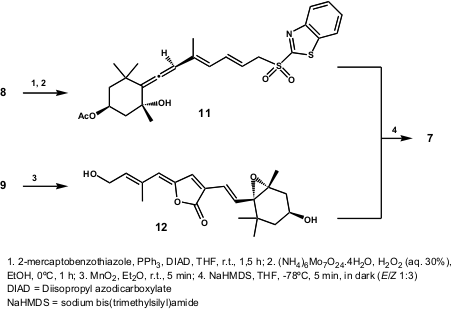
Scheme 3 – Total synthesis of Peridinin (7) by Katsumura and co-workers.
III. The Stille Reaction in the Synthesis of Carotenoids
Recently, the Stille reaction has been the method of choice for the synthesis of carotenoids as well as retinoids. The search for more efficient and convergent routes, which allow control over the olefin geometry (E/Z) under reaction conditions that are compatible with unstable and sensitive carotenoids, has led chemists to use the Stille cross-coupling reaction instead of the Julia or Wittig reactions.
Lera and co-workers have recently reported the synthesis of unsymmetrical carotenoids (Org. Lett. 2005, 7, 545.DOI: 10.1021/ol0478281)using the Stille reaction. The synthesis of 6´-epi-Peridinin (13) (Figure 2) was performed by this group where the usefulness of metal-catalyzed cross-coupling reactions in the synthesis of carotenoids was fully demonstrated. Scheme 4 shows the retrosynthetic approach developed by this group. The synthetic strategy was focused on two halogen-selective Stille reactions, one involving the dihalogenated fragment 14 with highly functionalized alkenylstannane 15 and the other between the resulting product and alkenylstannane 16. Haloallene 19 was chosen as a common synthon.
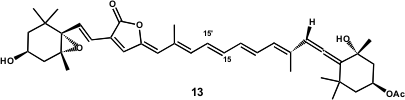
Figure 2 – 6´-epi-peridinin structure
The Lera group envisaged the synthesis of synthon 15 via a modified Julia Reaction of 17 with β-stannylacrolein and the formation of precursor 19 was also planned to employ a Stille reaction with an appropriated sulfone. Alkenylstannane 16 could be prepared from its precursor 18 by palladium-catalyzed hydrostannylation.
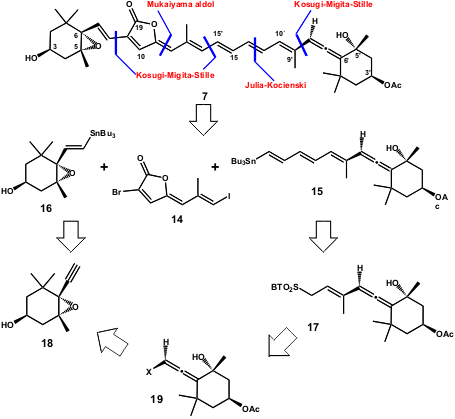
Scheme 4 – Retrosynthetic plan of Lera and co-workers for the synthesis of peridinin (7).
Nevertheless, when the Lera group used this methodology, they observed inversion of configuration at the allene centre of 17 during the cross-coupling (Scheme 5).
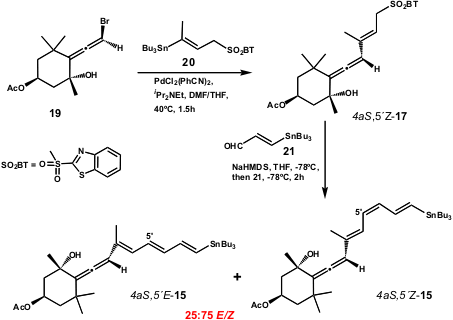
Scheme 5 – Inversion of configuration at the allene centre during the synthesis of allenyl allyl sulfone 17.
The authors interpreted this inversion as the consequence of an anti-selective SN2’-displacement of bromide by palladium, followed by a [1,3]-sigmatropic shift of propargyl to allenylpalladium which occurs before Stille reaction.
The use of Bu4N+ Ph2PO2– to shorten the reaction time was explained as due to the low reactivity of4aS,5´Z–17. The authors assigned the stereoselective coupling product’s structure as 13 (the 6´-epi–7) as the most stable E-isomer, as a result of the palladium induced isomerization of olefin. The total synthesis of 13was completed using an interesting and uniquely stereoselective Stille reaction of chiral enantiopure haloallenes with alkenylstannanes (Scheme 6).
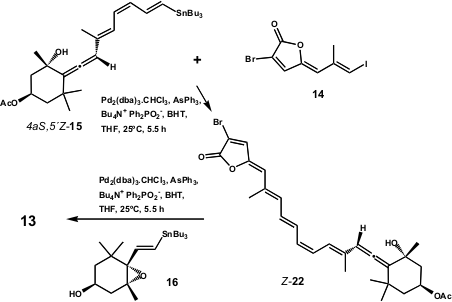
Scheme 6 – Total synthesis of 6´-epi-peridinin (13) by Lera and co-workers.
The same group had previously reported (J. Org. Chem. 2002, 67, 5040. DOI: 10.1021/jo025727f)that a similar strategy was applied to symmetrical carotenoids, where the Stille reaction was once more the method of choice.
IV. The Stille Reaction in the Synthesis of Retinoids
The Lera group also worked on the synthesis of retinoids (J. Org. Chem. 2000, 65, 5917.DOI: 10.1021/jo9917588), in particular some analogues of 9-cis-retinoic acid (23) (Figure 3).
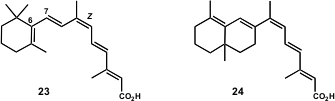
Figure 3 – Structures of 9-cis-retinoic acid (23) and the 6-s-trans-9-cis-retinoic acid analogue (24).
The retinoid 23 has the capacity of binding to RXRs, a subfamily of nuclear receptors, acting as modulators of nuclear transcription (Cell 1995,83, 835,DOI: 10.1016/0092-8674(95)90199-X;Cell1992, 68, 397,DOI: 10.1016/0092-8674(92)90479-V). Motivated by this interesting activity, the Lera group embarked on the synthesis of analogues of 23 with locked 6-s-cisand 6-s-trans conformations (C6-C7 bond, Figure 3), since the polyenic side chain of 23 plays an important role in the binding to RXR.
For the synthesis of 24, the Lera group planned a convergent and stereoselective approach via a Stille coupling (Scheme 7). The bicyclic dienyl triflate 25 and the (Z)-tributylstannylbut-2-en-1-ol (26) were used as starting materials, and were coupled under mild conditions. After oxidation of the resulting alcohol and subsequent Horner-Wadsworth-Emmons olefination followed by hydrolysis, the desired product was isolated in 77% yield.
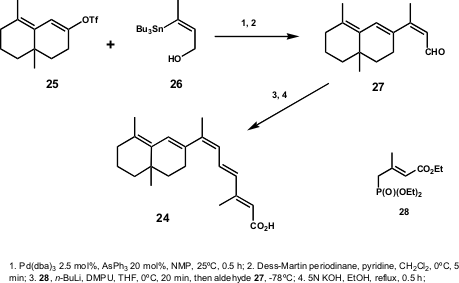
Scheme 7 – Total synthesis of the 6-s-trans-9-cis-retinoic acid analogue (24) developed by Lera and co-workers.
When Lera and co-workers tried an (E)-tributylstannylalkenol, they observed lower reactivity for the E-isomer as compared to Z-isomer in the Stille coupling. Thus, their study fully explored the reactions of stannylalkenols of different alkene geometry with different triflates, some highly steric hindered. They concluded that when an hydroxyl group is located cis to the carbon-tin bond, the observed rate difference between Z and E isomers is due to a coordination of palladium to the heteroatom in the transmetalation step, favouring Z-isomers (Scheme 8). However, the authors showed that the flexibility of this method is related to the E-isomer coupling product. Since the direct coupling is a low yield reaction in most cases, the same products can be prepared by isomerization of their Z-isomers at the carbonyl oxidation stage – 27 (Scheme 7).
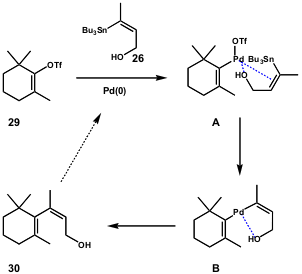
Scheme 8 – Model proposed by Lera and co-workers to explain the rate difference of Z– and E-isomers of alkenylstannanes.
The usefulness of the Stille reaction in the synthesis of the terpenoid skeleton of retinoids had been previously demonstrated by Lera and co-workers (Tetrahedron1999, 55, 15071.DOI: 10.1016/S0040-4020(99)00962-X).
Summary
Due to the important biological activity, olefin geometry and sensitivity of some of these compounds, the stereoselective synthesis of the polyene skeleton of carotenoids and retinoids is a demanding and challenging task. Recently, the search for efficient routes led to new strategies that involve the use of the Stille reaction. The choice of appropriate fragments for the coupling proves to be crucial to success.